
Avgörande kunskap om ovanlig hjärntumör hos barn



Pineoblastom är en sällsynt men mycket aggressiv hjärntumör som främst drabbar spädbarn och små barn. Ny forskning pekar på möjligheten till mer selektiv målstyrd behandling, riktad mot utvecklingsprogram som i stort sett saknar betydelse i normal vävnad efter tidig barndom, skriver professor Fredrik Swartling.
Trots intensifierad multimodal behandling är prognosen för pineoblastom fortsatt dålig. Under senare år har molekylär klassificering, framför allt via så kallad global metyleringsanalys förbättrat vår diagnostiska precision, men den biologiska förståelsen av tumörens ursprung och centrala drivmekanismer har varit begränsad. Vi har i en nyligen publicerad studie i vetenskapliga tidskriften Cancer Cell (Gudenas et al., 2026) studerat patientprover av pineoblastom och utvecklat en biologisk modell för hur pineoblastom uppstår och hur tumörformen relaterar till andra högriskdiagnoser inom barnonkologin. I denna översikt sammanfattar jag studiens huvudsakliga fynd och diskuterar deras kliniska relevans.
Cellursprung och utvecklingsfönster
Pineoblastom (PB) kan molekylärt delas in i fyra olika grupper: PB-miRNA1, PB-miRNA2, PB-MYC/FOXR2 och PB-RB1 som drivs av olika genetiska mekanismer och därför kan vara mer eller mindre elakartade (Liu et al., 2021). Pineoblastom som tillhör miRNA-grupperna drabbar oftast barn mellan 5-15 år, medan de sistnämnda grupperna drabbar yngre patienter (ofta spädbarn) och har sämre prognos med bara 30-35% överlevnad (Figur 1).
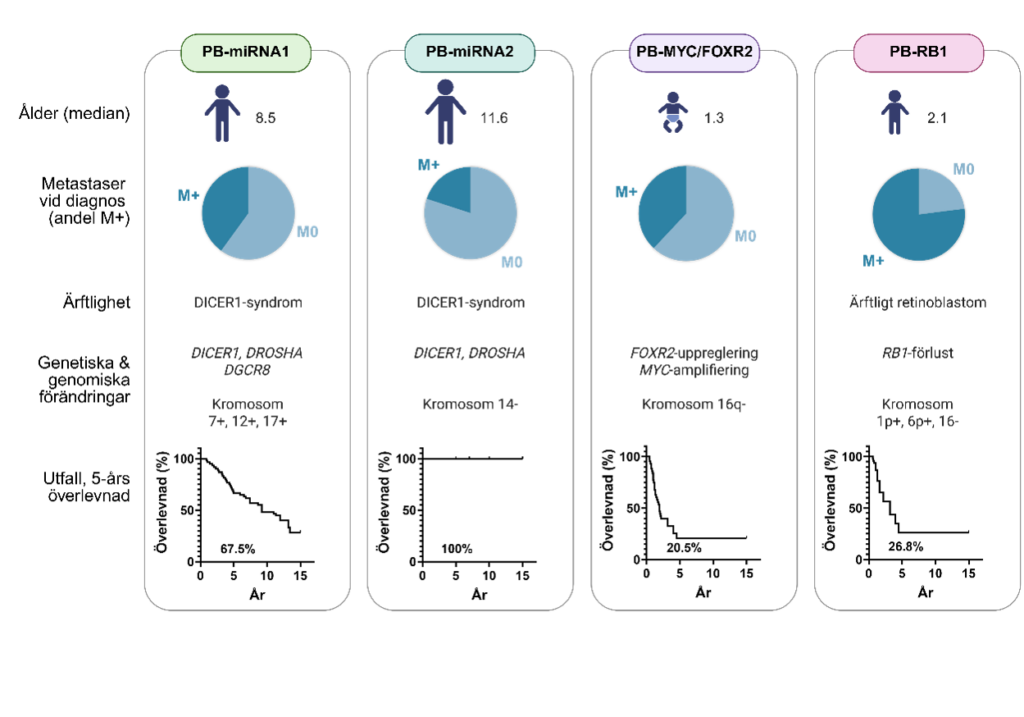
Figur 1. Schematisk översikt av den elakartade barnhjärntumören, pineoblastom utifrån patienter uppdelade i olika molekylära subgrupper. Medianålder beskrivs för insjuknande i de olika subgrupperna. M+ representerar pineoblastom vid diagnos där spridning av metastaser (andel fall i mörkblått) upptäcks i cerebrospinalvätskan eller i ryggmärgen, medan M0 (andel fall i ljusblått) är avsaknad av spridda tumörceller där. Ärftlighet och genetiska/genomiska förändringar kopplade till de olika grupperna anges om sådan finns. I procent anges 5-årsöverlevnad i de olika subgrupperna av pineoblastom. Figur ombearbetad från (Liu et al., 2021).
Pineoblastom tros härstamma från tallkottkörteln, ett endokrint organ som bland annat producerar melatonin, ett viktig hormon som reglerar kroppens vakenhet. Dess nivåer styrs av ljus som gör att vi effektivt kan hantera dag och natt i vår dygnsrytm. När näthinnan i ögat träffas av ljus går signaler till hypotalamus i hjärnan. Därifrån skickas stoppsignaler till tallkottkörteln att melatoninproduktionen ska upphöra. På kvällen när dagsljuset försvinner och vi förbereder oss för att sova, ökar utsöndringen av melatonin och vi blir sömniga.
För att förstå hur dessa tumörer uppstår genomförde vi enkelcells-RNA-sekvensering av ett större antal pineoblastom från opererade patienter i både USA och Europa. Tillsammans med två forskarteam ledda av Paul Northcott på St. Jude Children’s Research Hospital i Memphis och Mariella Filbin på Dana Farber, Harvard Univ. i Boston, USA visar vi att samtliga molekylära undergrupper av pineoblastom, oavsett genetisk drivare, uppvisar hög biologisk likhet med delande tidiga pinealocyt-progenitorer. Dessa celler representerar ett normalt, övergående utvecklingsstadium i tallkottkörteln och förekommer huvudsakligen under tidig postnatal utveckling.
Viktigt ur klinisk synvinkel är att tumörcellerna inte främst liknar omogna vuxna celler, utan snarare utvecklingsceller som behållit ett proliferativt program som normalt är strikt tidsbegränsat. Detta ger en biologisk förklaring till den tidiga debuten och den höga aggressiviteten.
Genetiska modeller
Studien stärks av funktionella data från genetiska musmodeller. Genom att inducera kända tumördrivande förändringar (till exempel RB1‑förlust eller MYC‑aktivering) selektivt i pinealocyt-linjen och under ett tidigt utvecklingsfönster kunde vi reproducera pineoblastom som utvecklades i djuren med hög specificitet. Samma genetiska ingrepp gav inte upphov till tumör när de aktiverades senare i livet. Detta understryker att tidpunkt och cellkontext är avgörande för tumörutveckling, ett koncept som har direkt bäring på hur vi tolkar genetiska fynd i kliniken.
Liknar andra högrisktumörer
Ett centralt fynd i studien är vår upptäckt av en gemensam molekylär profil mellan pineoblastom, retinoblastom och medulloblastom (av typ Grupp 3 som ofta har dålig prognos). Retinoblastom är en ögontumör hos barn som uppstår i just näthinnan (Zhou et al., 2024) (vars celler som nämnts ovan just har en koppling till tallkottkörteln genom att de skickar signaler efter att dagsljus träffar dem).
Medulloblastom tillhör den vanligaste typen av elakartade hjärntumörer hos barn (Hovestadt et al., 2020) (denna sjukdom drabbar cirka 15-20 barn varje år i Sverige). Med hjälp av enkelcells-sekvensering i kombination med flera molekylära metoder (inklusive global metyleringsanalys) hittade vi en signatur som är gemensam för dessa tre tumörtyper, benämnd tumor‑associated photoreceptor signature (TAPS), som domineras av gener som normalt reglerar fotoreceptorers utveckling och funktion, däribland CRX, OTX2 och NRL.
Vid jämförelse med ett stort spektrum av andra CNS‑tumörer framstår TAPS som i stort sett unik för dessa tre diagnoser. Detta tyder på att tumörerna delar ett gemensamt utvecklingsberoende trots olika anatomisk lokalisation (Figur 2).
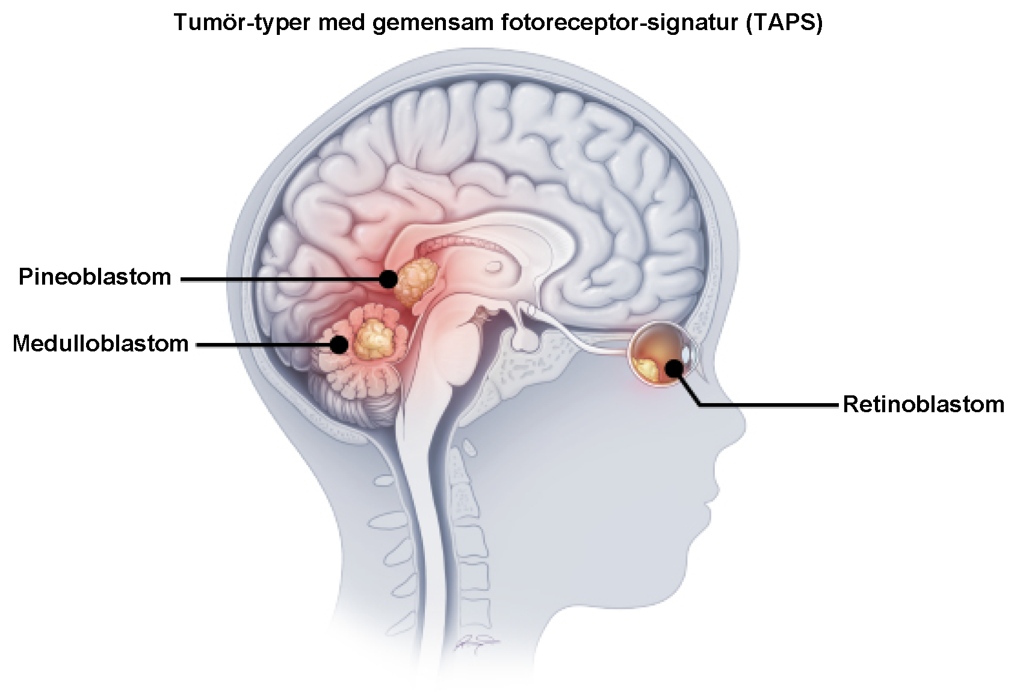
Figur 2. Trots sin lokalisation i tre helt skilda regioner i hjärnan visar tumörceller från pineoblastom, medulloblastom och retinoblastom en gemensam molekylär signatur för fotoreceptorer (TAPS) som uppstår i begränsat tidsfönster under olika delar av hjärnutvecklingen. Tumörerna är beroende av denna TAPS-signatur vilket möjliggör att dessa tumörtyper potentiellt kan behandlas på ett liknande sätt i framtiden. Bilden är något ombearbetad och hämtad från (Gudenas et al., 2026).
Implikationer för framtida behandlingar
Av särskilt kliniskt intresse är att flera nyckelkomponenter i TAPS visade sig vara funktionellt nödvändiga för tumörcellernas överlevnad. CRISPR‑baserade studier visade att nedreglering av centrala delar av detta program hämmade tumörtillväxt i modeller av pineoblastom, retinoblastom och Grupp 3‑medulloblastom, men inte i andra hjärntumörer.
Detta pekar på möjligheten till mer selektiv målstyrd behandling, riktad mot utvecklingsprogram som i stort sett saknar betydelse i normal vävnad efter tidig barndom. Om det lyckas kan neuronala och kognitiva biverkningar som ofta drabbar dessa barn begränsas.
Klinisk relevans och framtida perspektiv
Sammanfattningsvis stärker studien bilden av hjärncancer som en sjukdom där normala utvecklingsprogram aktiveras i fel sammanhang (antingen sker detta spontant av slumpmässiga mutationer eller som uppstår på grund av en ökad risk från en bakomliggande ärftlig sjukdom (se exempel i Figur 1)). För barnonkologin innebär vår upptäckt att aggressiva tumörer som tidigare betraktats som biologiskt disparata nu kan förklaras och beskrivas inom ett gemensamt ramverk. På sikt kan detta få konsekvenser för diagnostik, riskstratifiering och terapiutveckling för barn som drabbas av dessa allvarliga sjukdomar.
Pineoblastom och dess ursprung
Pineoblastom (PB)
- Sällsynt, höggradig barnhjärntumör i tallkottkörteln
- Debuterar oftast innan 5 års ålder
- Frekvent spridning i cerebrospinalvätskan vid diagnos
- Låg långtidöverlevnad trots intensiv behandling
Molekylära egenskaper hos PB
- RB1‑associerade ofta ärftliga tumörer
- MYC‑driven undergrupp
- MikroRNA‑processeringsdefekter
- Gemensamt: hög proliferation och embryonal genexpression
Nyckelfynd i aktuell studie
- Tumörerna härstammar från delande pinealocytprogenitorer.
- Tumörutveckling kräver ett specifikt tidigt utvecklingsfönster.
- Gemensam fotoreceptorliknande transkriptionssignatur (TAPS) delas med:
Retinoblastom
Medulloblastom av typ Grupp 3
Klinisk betydelse av aktuell studie
- Fördjupad biologisk förståelse av högrisktumörer hos barn.
- Identifierar potentiella nya terapimål.
- Stödjer utvecklingsbaserad klassificering av CNS‑tumörer.

Missa inte artikeln Patientnytta ska prioriteras – utlysning öppnar i juni och Ny strategi mot svårbehandlad barncancer
Referenser
Gudenas, B.L., Ahmad, S.T., Englinger, B., Liu, A.P.Y., Zhao, M., Paul, L., Hadley, J., Li, Y., Batts, M., Mittal, P., et al. (2026). A tumor-associated photoreceptor signature unifies distinct central nervous system malignancies. Cancer Cell. 10.1016/j.ccell.2026.02.010.
Hovestadt, V., Ayrault, O., Swartling, F.J., Robinson, G.W., Pfister, S.M., and Northcott, P.A. (2020). Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat Rev Cancer 20, 42-56. 10.1038/s41568-019-0223-8.
Liu, A.P.Y., Li, B.K., Pfaff, E., Gudenas, B., Vasiljevic, A., Orr, B.A., Dufour, C., Snuderl, M., Karajannis, M.A., Rosenblum, M.K., et al. (2021). Clinical and molecular heterogeneity of pineal parenchymal tumors: a consensus study. Acta Neuropathol 141, 771-785. 10.1007/s00401-021-02284-5.
Zhou, M., Tang, J., Fan, J., Wen, X., Shen, J., Jia, R., Chai, P., and Fan, X. (2024). Recent progress in retinoblastoma: Pathogenesis, presentation, diagnosis and management. Asia Pac J Ophthalmol (Phila) 13, 100058. 10.1016/j.apjo.2024.100058.



